



 01
01Previously, we’ve discussed what life’s chemical code is, why we built the first “sequencer” technology for this chemical code, how we are first directing this sequencer at traditionally used medicinal plants to find new medicines, and the value of this approach for valuing biodiversity.
The chemistry that underlies our approach—the evolved small molecules generated by and for biology—has another key benefit: it has allowed us to create a platform on which we can execute drug discovery and new target discovery in parallel and at scale.
Typically, in pharma, target discovery and molecule discovery are seen as separate and distinct. We are pioneering a new approach that harmonizes these two currently independent processes into an efficient, unified process, revealing both new biology and the new chemistry with which to prosecute it.
There’s historical precedent for the idea that nature’s chemistry can reveal new insights into biology and new drug targets, with aspirin leading to the discovery of COX enzymes and rapamycin revealing the fabled TOR pathway. However, what’s been missing until now is speed and scale.
In this post, I will discuss the key attributes of our library and platform that allow us to synchronize these processes and how this has resulted in multiple developmental candidates in just the 4 years since our company’s founding.
02The power behind our approach is our library, which has two key features: high diversity and high relevance. This work could not be carried out with the standard synthetic libraries currently used by most pharmaceutical companies for screening; for example, interrogation of TNFa signaling using standard synthetic libraries would only reveal known targets, largely kinases.
We define high relevance by two axes: the first is relevance to biology. Synthetic libraries are constrained by what humans can synthesize in the laboratory. Our library is constrained by evolution itself, representing billions of years of selective pressures on the molecular arrangements most conducive to interacting with, being shaped by, and even shaping biological pathways.
The second axis is relevance to disease. Our library is composed of molecules derived from medicinal plants that, in some cases, have centuries of prior use demonstrating their potential utility in treating some of our most complex diseases. As discussed in this publication, we use advanced data science methodologies to map patterns of medicinal plant use across cultures, and across time. With this map, we can identify with confidence the plants that are most likely to contain molecules that will interact with disease-relevant pathways and influence health outcomes, which we then source and incorporate into our library.
We define high diversity across all quantitative chemical characteristics. Below we show this with one graph that encompasses many of these characteristics.
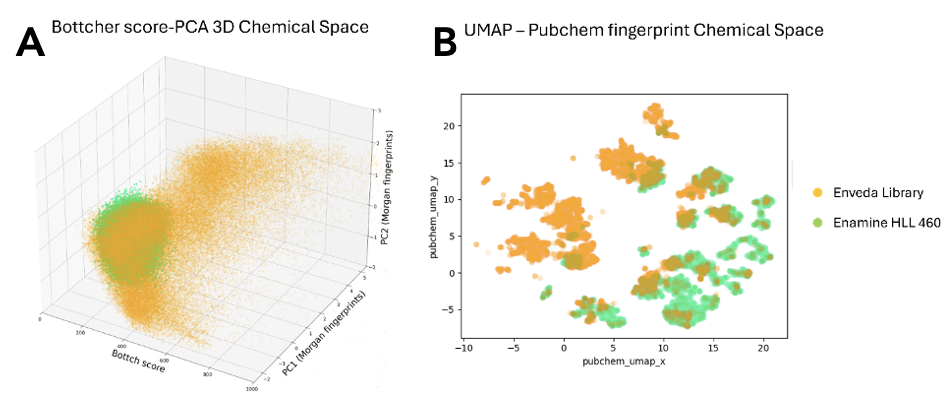
Figure 1. The chemical diversity contained in Enveda’s natural product library far exceeds that of synthetic libraries. The graphs above compare a random sampling of 90k compounds from either Enveda’s library or the Enamine Hit Locator Library 460 (chosen because it covers the entire chemical diversity of Enamine’s expanded collection). On the left (A), we compare these two sample sets by 3D chemical diversity using the Bottcher score and two additional PCA analyses using Morgan Fingerprints. On the right (B) is a 2D UMAP using PubChem Fingerprints. In both examples, Enveda’s library in orange covers a larger, unique chemical space that compliments a typical synthetic library.
In summary, our library lets us start our drug discovery process with unparalleled structural diversity, that is the result of evolutionary selection for highly conserved biology, and from plants with precedence for utility. By screening with a diverse and highly relevant library, we are able to obtain higher quality hits that have higher chances of making it through the battery of subsequent tests before successfully becoming a drug.
03As Viswa describes in his blog post, one of the main reasons why exploring life’s chemistry was (until Enveda) such a difficult and slow process is because it had to be done one molecule at a time. Scientists would isolate a single molecule from a natural source and then painstakingly determine its structure and function. To be able to truly capture and utilize the wisdom represented in life’s chemistry, we had to scale this process, which meant that we had to develop a platform that could deduce a novel molecule’s structure and function while still within a complex mixture.
To do this, we screen our extract mixtures in high throughput, 1536-well bioactivity, and binding assays using the same setup as a pure compound screen. We first collect the MS2 (or mass spectra) for each compound in each well. The pattern represented in an MS2 is unique to each molecule, and we are able to sample 100s of MS2s in seconds. The MS2s are then used much like a fingerprint to track compound presence in particular wells. If you’ve read our blog posts on MS2Mol and MS2Prop, you’ll know that we also use the MS2 to predict the structure and, thus, the identity of the molecules. In this way, we’re using a single datatype as both a unique tag and as an answer to the most important question medicinal chemists have about a potential lead. Simultaneously with this structure prediction, we are running each sample through a battery of biological assays, which I will go into more detail on below. By being able to identify molecules and ascribe structure and functional information to them while still in a mixture, we circumvent the need to do painstaking isolations at the initial stage. Instead, we only do isolations after we have compelling evidence that a molecule in question has the desired activity profile and chemical amenability necessary to be able to optimize it with medicinal chemistry. This step is crucial in integrating the library of life with modern drug discovery at an unprecedented scale.
04It may seem antithetical that the basis of our approach to combining target discovery and drug discovery is phenotypic screening. Phenotypic screening has gone in and out of vogue in pharma, with the major critique leveled against it being that you run a high risk of rediscovering known (and possibly undesirable) targets. This concern makes sense if you are using standard synthetic libraries. Given their lack of diversity, there is a finite number of proteins, protein complexes, or other macromolecules (and their complexes) that these compounds will interact with. Moreover, their mode of interaction and the resulting bioactivity is likely constrained to enzymatic inhibition. Our library, on the other hand, is likely both the most diverse and most relevant chemistry for drug discovery. Even in standard, constrained, cell-based signaling assays, most of our hits do not recapitulate known biology. When used in combination with new tools to quickly and reliably deconvolute the mechanism of action (MOA), we can provide swift validation of target identity. With these considerations, we’re able to minimize the downsides of phenotypic screening so that we can maximize its key upside: the opportunity to discover new disease biology, and thus drug targets. To further the above example, a TNFa signaling assay at Enveda will yield hundreds of specific hits but very few Jak inhibitors.
Since our library is enriched in these highly diverse, bioactive natural molecules, we can use them both as chemical probes to help us determine new targets and new MOAs and as starting points for first-in-class discovery. We employ this powerful library in phenotypic and pathway activity screens. The beauty of these screens is that they allow us to select for the biological effect we want to elicit but without constraining how a therapeutic molecule exerts that effect, opening up the scope of possible solutions.
To illustrate this process, let’s walk through one example. There is long-standing evidence that a class of medicinal plants exert anti-inflammatory effects. To identify the anti-inflammatory molecules within these plants, we perform phenotypic screens for molecules that induce anti-inflammatory effects, as well as pathway activity screens for the different cytokine pathways we may want to modulate, allowing us to find inhibitors of the receptor/ligand interaction, but also inhibitors of any one of the known or unknown proteins that lie upstream or downstream of these signal transduction pathway. Once a hit is identified, we use a combination of our metabolomics expertise and modern chemoproteomic approaches to rapidly identify the target protein for a molecule of interest. We also leverage our other platform technologies, such as MS2Mol, to ensure that our candidate molecules are accessible and amenable to medicinal chemistry. Lastly, we perform target-centric screens to refine and optimize our candidates. This also allows us to leverage the diversity of our library to prosecute screens on well-characterized targets.
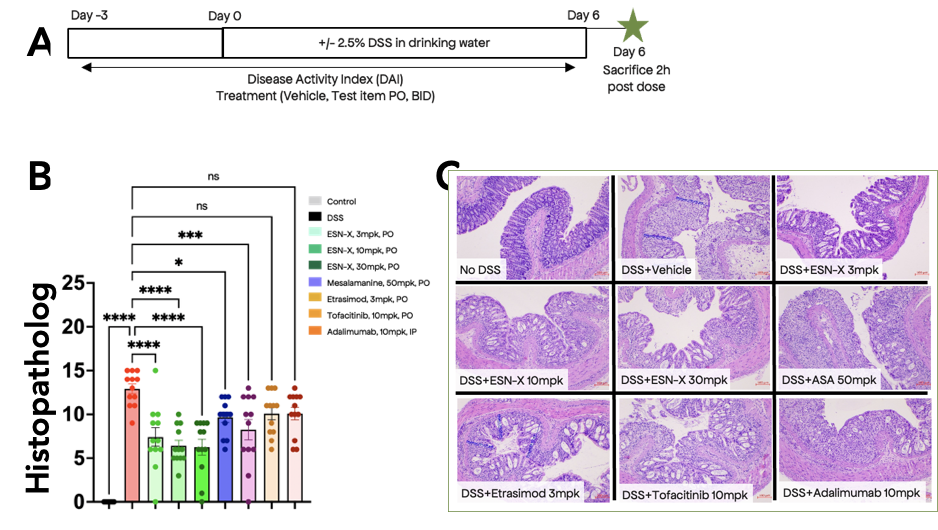
Figure 2. ESN-X outperforms current standards of care in the DSS mouse model of Inflammatory Bowel Disease. Our benchmarking studies utilized a 6 day treatment regime to induce colitis (A) and efficacy was measured by a composite score of multiple disease relevant readouts (B) and permeability of the gut (C).
A perfect example of how our approach has led us to novel biology is the discovery process that resulted in our second candidate, ESN-X, for the treatment of inflammatory bowel disease. This condition is characterized by gut inflammation, which can occur either in the lining of the digestive tract or throughout the large intestine, colon, and rectum. Multiple inflammatory pathways have been validated through the successful development of biologics (against TNFa, IL23, and TL1A) , but despite targeted approaches of many drug discovery programs, there remains a high unmet need. Without giving away too much, ESN-X is derived from a medicinal plant with a long history of use as an anti-inflammatory, including symptoms of inflammatory bowel disease. We first identified the original hit in a pathway screen looking for inhibitors of the NLRP3 inflammasome pathway. Once we identified the molecule driving the anti-inflammatory activity, target deconvolution allowed us to pinpoint a novel mechanism of targeting inflammasome activation, degrading the target NLRP3 protein via a first-in-pathway target, unlike the previous generation of direct NLRP3 inhibitors. Moreover, this mechanism suggested we could have effects in other key inflammatory pathways. Indeed, on further testing in screens for inhibition of additional cytokine pathways, we identified that our molecule exerts anti-inflammatory effects across other inflammatory pathways, such as TL1A and IL23, with a compelling molecular mechanism. In preclinical studies, ESN-X outperforms multiple existing approved therapeutics (figure 2), including S1P1s and Jak-inhibitors. We believe, and have strong preclinical data to support, that this will result in a more complete resolution of inflammation for those who suffer from inflammatory bowel disease while still maintaining systemic immune system function. As a biologist with a deep-rooted urge for discovery, I am in awe at how beautifully this story has unfolded, consistent with the core thesis of Enveda. I hope this is one of many ways in which we deliver on the opportunity to identify novel biological targets leading to a new understanding of immune regulation.
05With our success in industrializing target and molecule discovery, we hope to deliver novel, differentiated, first-in-class therapies for complex diseases with large medical need. How many more mTOR pathways does Nature have yet to teach us about?